DFT Caluclations
The crystal structure of pMMO reveals that the protein is broken into three subunits, in each subunit lies three metal centers. Two of these metal centers (a mononuclear and a binuclear copper site) are located within the soluble regions of the pmoB subunits. The third metal site is modeled to have a Zinc center.
The identity of the metals at the active site is still debated between mixtures of Fe, Zn, and Cu active metals. Recently it has been shown that the activity of the protein is greatly enhanced when Copper is added to the growth medium. This implies that Copper could play an essential role in the activity of the protein. However, the oxidation states of the Copper is widely debated.
Recently, Yoshizawa decided to determine the reactivity of the active site, based off the recently obtained crystal structure. He modeled the mono-copper site as a Cu(III)-oxo site coordinated to two Histidine and a Glutamate. The overall charge is neutral owing to the Glu ligand. The Di-copper site is modeled to be bis(m-oxo)Cu(II)Cu(III) with three His and Glu coordinating to the site.
The identity of the metals at the active site is still debated between mixtures of Fe, Zn, and Cu active metals. Recently it has been shown that the activity of the protein is greatly enhanced when Copper is added to the growth medium. This implies that Copper could play an essential role in the activity of the protein. However, the oxidation states of the Copper is widely debated.
Recently, Yoshizawa decided to determine the reactivity of the active site, based off the recently obtained crystal structure. He modeled the mono-copper site as a Cu(III)-oxo site coordinated to two Histidine and a Glutamate. The overall charge is neutral owing to the Glu ligand. The Di-copper site is modeled to be bis(m-oxo)Cu(II)Cu(III) with three His and Glu coordinating to the site.
Optimized Structures of Various Metal Sites
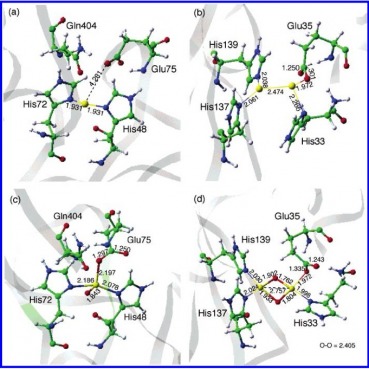
This figure contains the optimized structures from the QM/MM calculations concerning the active metal sites. (a) being the mono-copper site. (b) the di-copper site. (c) the mono-copper oxo species. (d) the di-copper oxo species. based upon the proposed oxidation states listed above. Using these optimized conditions, they calculated the energy states using the B3LYP* level of theory.
Energy Diagram for the Mono-Copper Site.
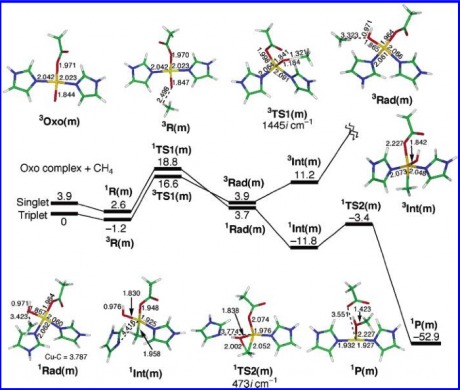
The model begins with the Copper species already in the oxo-form. The first intermediate (R(m)) is the one is which methane approaches the active site and is loosely bound the the active site. The methane is directed to the active site via a hydrophobic pocket contained within the protein. This first intermediate is slightly lower in energy than the preceding species.
The next step is to the TS1(m) state. This is the most energetically demanding transition because it is the one in which there is the homolytic cleavage of the C-H bond via the radical on oxygen. This transition state leads to the formation of the radical intermediate Rad(m). Also based of the energies, this is where the expected switch from a triplet to singlet spin state would occur.
This Radical intermediate quickly gives way to a more stable non-radical intermediate Int(m). The second transition state (TS2(m)) is not a terribly uphill reaction, certainly less than the first reaction state. The second transition state being a triangular structure that allows for the recombination of OH with CH3.
Thus the RDS is the radical cleavage of the CH bond, with the overall reaction process being about 52.9 kcal/mol downhill.
The next step is to the TS1(m) state. This is the most energetically demanding transition because it is the one in which there is the homolytic cleavage of the C-H bond via the radical on oxygen. This transition state leads to the formation of the radical intermediate Rad(m). Also based of the energies, this is where the expected switch from a triplet to singlet spin state would occur.
This Radical intermediate quickly gives way to a more stable non-radical intermediate Int(m). The second transition state (TS2(m)) is not a terribly uphill reaction, certainly less than the first reaction state. The second transition state being a triangular structure that allows for the recombination of OH with CH3.
Thus the RDS is the radical cleavage of the CH bond, with the overall reaction process being about 52.9 kcal/mol downhill.
Energy Diagram for the Di-Copper Site
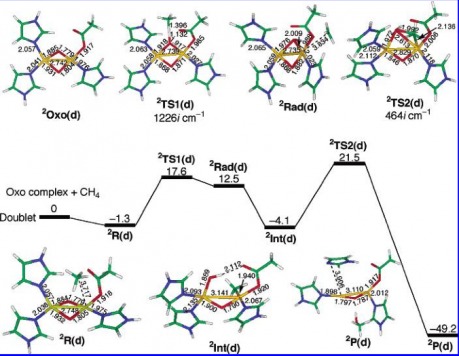
Once again the methane is weakly bound to the active site, specifically the right-side copper. After this, the CH bond is homolytically cleaved via a free radical from the oxygen. This forms the first transition state TS1.
This leaves the Radical trapped on the right side copper, which again will form the energetically favored non-radical intermediate. This is going from methyl radical Rad(d) to the non-radical intermediate Int(d).
The final step is the formation of TS2(d) which is the three-membered ring containing the Copper, the OH, and the CH3 fragment. In the case of the di-copper site, the second transition state is the RDS.
The overall transformation is favored by 49.2 kcal/mol.
This leaves the Radical trapped on the right side copper, which again will form the energetically favored non-radical intermediate. This is going from methyl radical Rad(d) to the non-radical intermediate Int(d).
The final step is the formation of TS2(d) which is the three-membered ring containing the Copper, the OH, and the CH3 fragment. In the case of the di-copper site, the second transition state is the RDS.
The overall transformation is favored by 49.2 kcal/mol.
Energy of Formation of Oxygen Species
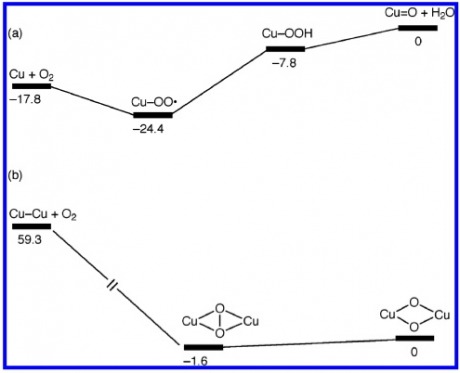
For the mono-copper site we can see that the formation begins with oxygen incorporating into the Cu(I) species to yield the Cu(II) superoxo. This is followed by an H-atom transfer from a local residue. (most likely from a distal Tyrosine). The addition of another proton yields the Cu(III)-oxo species plus an equivalent of water. This required 17.8 kcal/mol of energy which is possible under physiological conditions.
The Di-copper site has insertion of oxygen to form a peroxo compound. This is transformed into bis(m-oxo)Cu(II)Cu(III). This active site is actually 59.3 kcal/mol in the favorable direction, making this a promising active site from which the reaction occurs.
The Di-copper site has insertion of oxygen to form a peroxo compound. This is transformed into bis(m-oxo)Cu(II)Cu(III). This active site is actually 59.3 kcal/mol in the favorable direction, making this a promising active site from which the reaction occurs.
Conclusions
The calculations that where done showed that the formation of methanol from methane were possible under physiological conditions using copper exclusively at the active site of the molecule. The mono-copper site showed a more favorable formation energetically from the oxo-form. However, the oxo-form was formed more readily in the di-copper site than in the mono-copper site. This shows that in the overall transformation, the di-copper site should be favored energetically.
Citations
Lieberman, R. L.; Rosenzweig, A. C. Nature 2005, 434, 177.
Yoshizawa, K.; Shiota, Y. J. Am. Chem. Soc. 2006, 128, 9873-9881.*
Yuan, H.; Collins, M. L. P.; Antholine, W. E. J. Am. Chem. Soc. 1997, 119, 5073.
Elliot, S. J.; Zhu, M.; Tso, L.; Nguyen, H.-H. T.; Yip, J. H-K.; Chan, S. I. J. Am. Chem. Soc. 1997, 119, 9949.
*All Figures where obtained from this paper.
Yoshizawa, K.; Shiota, Y. J. Am. Chem. Soc. 2006, 128, 9873-9881.*
Yuan, H.; Collins, M. L. P.; Antholine, W. E. J. Am. Chem. Soc. 1997, 119, 5073.
Elliot, S. J.; Zhu, M.; Tso, L.; Nguyen, H.-H. T.; Yip, J. H-K.; Chan, S. I. J. Am. Chem. Soc. 1997, 119, 9949.
*All Figures where obtained from this paper.
